Viral Vector Long‑Read NGS
Understand impurities early. See what standard assays miss. De-risk you vector design.



Understand impurities early. See what standard assays miss. De-risk you vector design.

Early‑stage gene therapy programs often move forward without a full understanding of what’s actually inside the vector. Truncations, backbone carryover, rep/cap fragments, host‑cell DNA, or other unexpected species can remain invisible until much later.
By that point, fixing the problem is expensive, time‑consuming, and can jeopardize regulatory submissions or even clinical safety.
Discovery teams need a way to uncover these hidden impurities early, during vector optimization and candidate selection, when design decisions are still flexible and inexpensive to change.

Our long‑read nanopore NGS service provides a complete, unbiased view of your vector—capturing every DNA species present, including unexpected impurities that targeted assays miss.
With extremely low material requirements (e.g. as little as ~5E9 vg for AAV), it’s ideally suited for groups who want deep characterization without needing to manufacture high‑titer batches.
Whether you’re selecting between multiple candidates, validating a novel production platform, or de‑risking your plasmid system, our analysis gives you confidence that your vector payload is structurally sound and ready for the next stage.

Sequence length distribution reveals the proportion of full-length vector genomes relative to shorter fragments, enabling detection of truncations and structural heterogeneity. The dominant peak corresponding to the expected genome size indicates intact packaging, while absence of secondary peaks supports minimal truncated species. Optionally, read size profiles can be corrected for Oxford Nanopore sequencing size bias with co-sequenced lambda phage DNA digest.
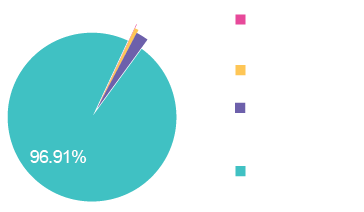
NGS reads are mapped against reference sequences to quantify contributions from the vector genome, plasmid backbone components, helper elements, and host cell DNA. This analysis provides a comprehensive view of sample composition and enables detection of residual process-related impurities not captured by targeted assays.
Coverage plots show sequencing read distribution across the ITR-to-ITR region, including promoter, transgene, and regulatory elements, as well as nucleotide polymorphisms. Uniform coverage supports intact genome representation, while localized drops or discontinuities may indicate structural abnormalities detectable through NGS.
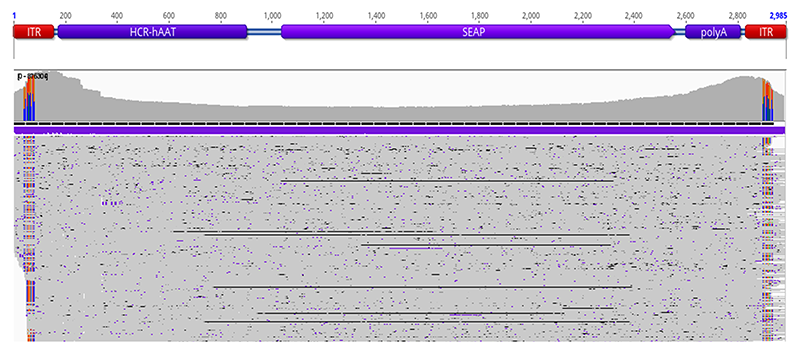
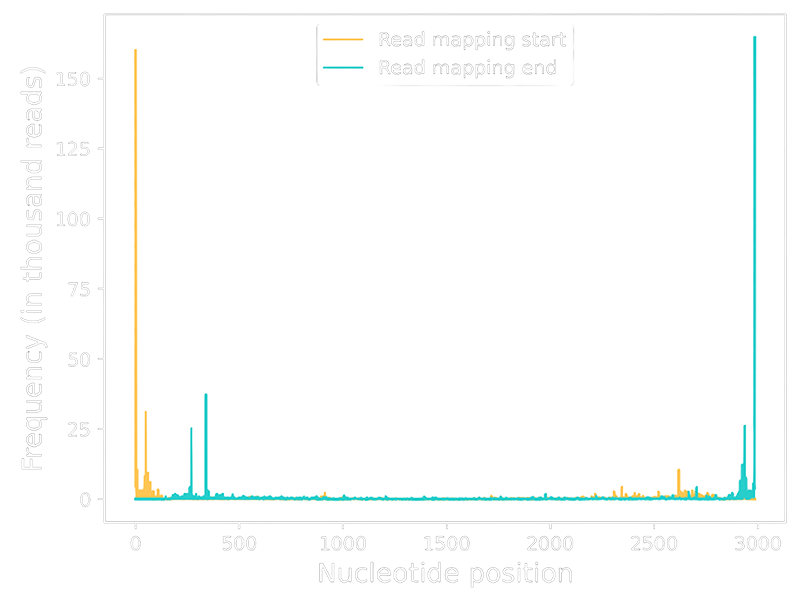
Base-level analysis enables identification of truncation events across the vector genome, allowing early identification of potential quality or design risks.
Typical material requirements for AAV:
Modality support:
What our experts deliver:
Please contact us using the form below.
"*" indicates required fields
Recombinant AAV batch profiling by nanopore sequencing elucidates product-related DNA impurities and vector genome length distribution
Advancements in nanopore sequencing allow in-depth characterization of rAAV vector batches comparable to SMRT™ sequencing
The Good, the Bad, & the Chimeric: Using Nanopore Sequencing to De-Risk rAAV Genome Packaging
Recombinant AAV batch profiling by nanopore sequencing elucidates product-related DNA impurities and vector genome length distribution
Advancements in nanopore sequencing allow in-depth characterization of rAAV vector batches comparable to SMRT™ sequencing
The Good, the Bad, & the Chimeric: Using Nanopore Sequencing to De-Risk rAAV Genome Packaging
